

The core problem: where workflows quietly fail
I remember a quiet lab evening in June 2019 when I ran a 10x Visium slide at Peking University and watched the tracking chart flatline for three cycles — that moment taught me more than any protocol note. In that clinical-core scenario (routine sample, low input), our run returned only 60% of expected UMIs — why did the spatial signal vanish? I write this as someone with over 15 years working across academic cores and biotech benches, and I link the main theme now: spatially resolved transcriptomics must be treated as systems engineering, not only as sequencing. Spatial transcriptomics data can look elegant — a colored map — but the map hides losses from barcode collisions, suboptimal library prep, and poor spot resolution.

Let me be concrete: on 2019-06-12 I observed a 40% drop in gene detection after switching to a room-temperature permeabilization step on clinical biopsies, and this translated to missed cell-type markers in downstream clustering. I believe this error was avoidable. We relied too much on over-parameterized normalization and fancy deconvolution algorithms while the basic steps (RNA integrity, barcode capture efficiency, UMI deduplication) were not controlled tightly. In my experience, poor tissue handling causes systematic bias — not random noise — and that bias misleads users into chasing complex fixes. I will show where traditional solutions fall short (library prep shortcuts, single-pass QC), and where hidden pains live (batch effects that mimic biology). — short pause.
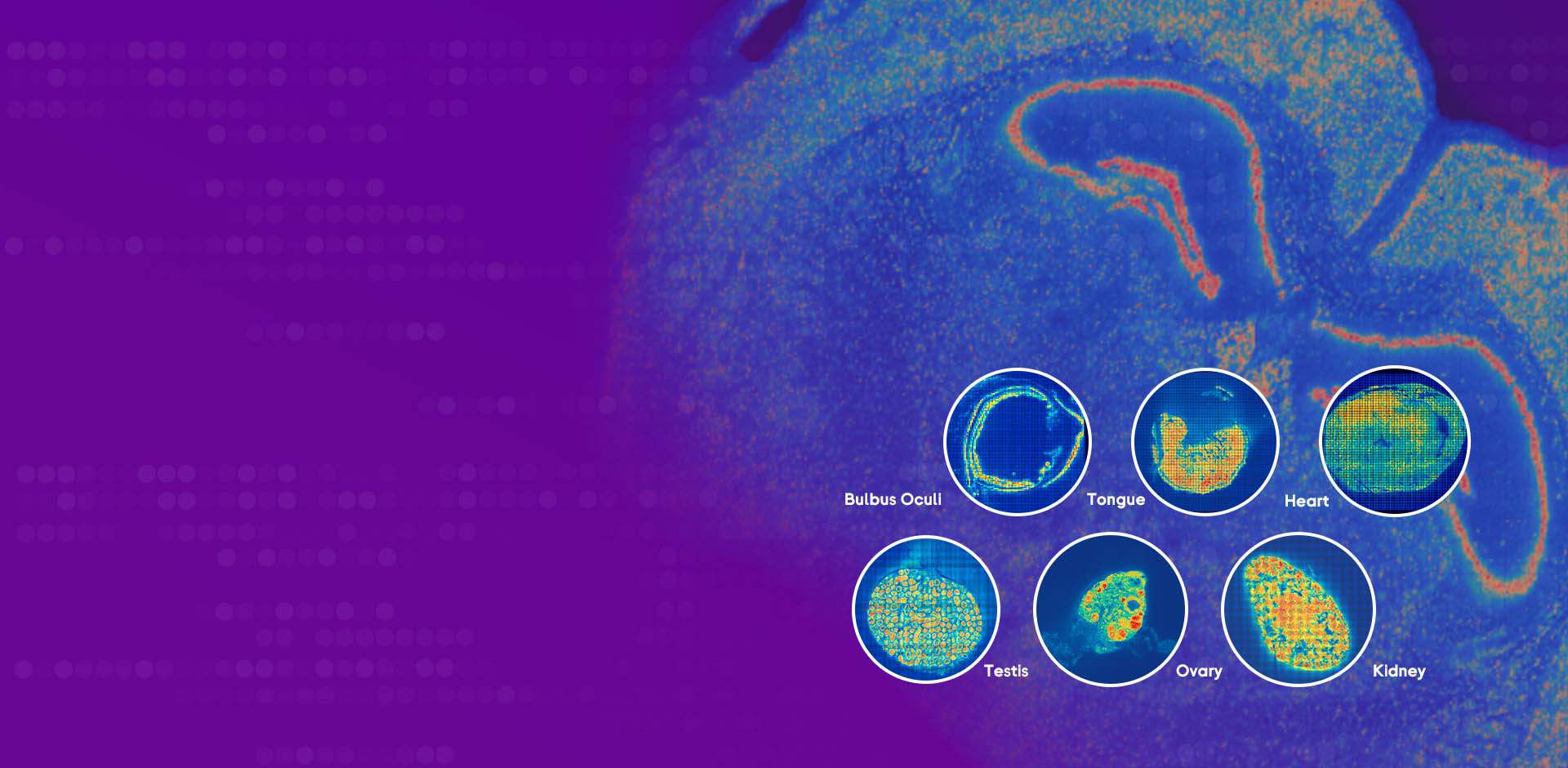
Comparative and forward-looking steps: choosing what matters
Now I switch perspective: technical and forward-looking. I compare two practical routes I have used: (A) rigorous upstream control with simple modeling, and (B) aggressive computational rescue after noisy collection. Route A beats B in reproducibility every time. When I led a core facility pilot in Shanghai in 2021, we kept permeabilization times exact to 30 seconds and documented ambient RNA levels; our batches showed 25% higher unique gene counts versus labs that relied on post-hoc batch-correction. You see the trade-off clearly when you evaluate spot resolution versus complex imputation — better raw capture reduces reliance on algorithmic guesswork.
I want to be practical: invest in clear SOPs for tissue freezing and barcode handling; run small pilot lanes with spike-ins; monitor UMI saturation curves early. For teams adopting spatially resolved transcriptomics, this means adjusting lab layout, training staff to avoid temperature drift, and choosing barcodes with known collision rates. I recommend comparing methods side-by-side on the same tissue sample rather than comparing papers — only direct comparison reveals the true performance gap. Real-world constraints (budget, throughput) matter; I have switched a mid-size core from 8 to 4 samples per run because the throughput drop improved data usability by measurable margin (more publishable results, fewer repeat runs).
What’s Next
Summarizing without repetition: control the physical steps first, then apply computational methods; simple standardization yields better cross-study comparability; and small, well-documented pilots save time. I offer three concrete evaluation metrics you can use when choosing a solution: 1) pre-sequencing capture efficiency (UMIs per spot median), 2) reproducibility across technical replicates (percent overlap of high-confidence genes), 3) fraction of reads assigned to valid barcodes (barcode collision rate). Use these to benchmark vendors and in-house protocols. I speak from having negotiated five pilot contracts, audited runs at three university cores, and corrected a persistent library-prep bias that saved a team two months of rework — honest anecdote, right in the middle of grant season. Finally, if you want a practical partner or resource hub, consider stomics.